[Source: David Orenstein | The Picower Institute for Learning and Memory | April14, 2026]
Using advanced human cell cultures to model Rett syndrome, MIT researchers tracked how two different mutations alter neural circuit development and how each could be addressed with distinct potential therapeutics
Though many studies approach the developmental disorder Rett syndrome as a single condition arising from general loss of function in the gene MECP2, a new study by neuroscientists in The Picower Institute for Learning and Memory at MIT shows that two different mutations of the gene caused many distinct abnormalities in lab cultures. Moreover, correcting key differences made by each mutation required different treatments.
“Individual mutations matter,” said Mriganka Sur, senior author of the new study in Nature Communications and Newton Professor in The Picower Institute and the Department of Brain and Cognitive Sciences. “This is an approach to personalizing treatment, even for a single-gene disorder.”
The study employed advanced 3D human brain tissue cultures called “organoids” or “minibrains” derived from skin cells or blood cells donated by Rett syndrome patients with each mutation. Lead author Tatsuya Osaki, a Picower Institute research scientist, said that the organoids’ ability to model the specific consequences of each mutation enabled him to gain mutation-specific insights that haven’t emerged in prior studies where scientists have just knocked out MECP2 overall. The organoids also provided a novel opportunity to understand how each mutation affected different cell types and their interactions.
Distinct effects
More than 800 mutations in MECP2 can cause Rett syndrome, but just eight account for more than 60 percent of cases. Sur and Osaki chose one of these, R306C, which involves a difference of just one DNA base pair (916C>T), because it represents 7-8 percent of Rett syndrome cases. The other mutation they chose, V247X, is much more rare and severe because it cuts off production of the gene’s protein product by a single DNA base deletion (705Gdel), leaving the protein not just errant, but incomplete.
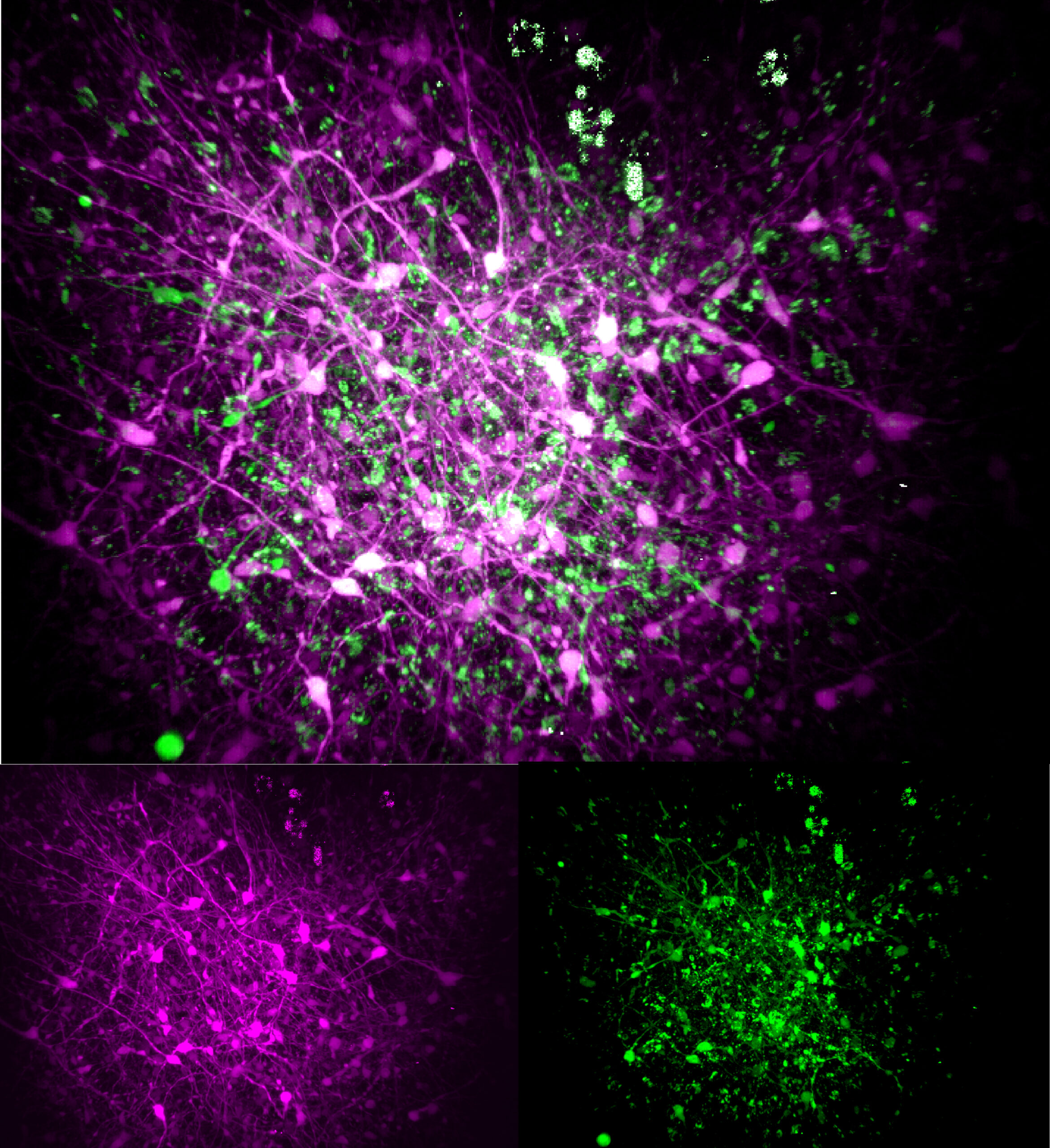
In organoids cultured for three months, each mutation produced some common but also sometimes distinct consequences compared to control organoids with non-mutated MECP2. For many of their experiments, the team used “three-photon” microscopes capable of cellular-level resolution all the way through the organoids’ ~1mm thickness, resolving both their structure (via “third-harmonic generation” imaging), and the live activity patterns of their neurons (via calcium fluorescence).
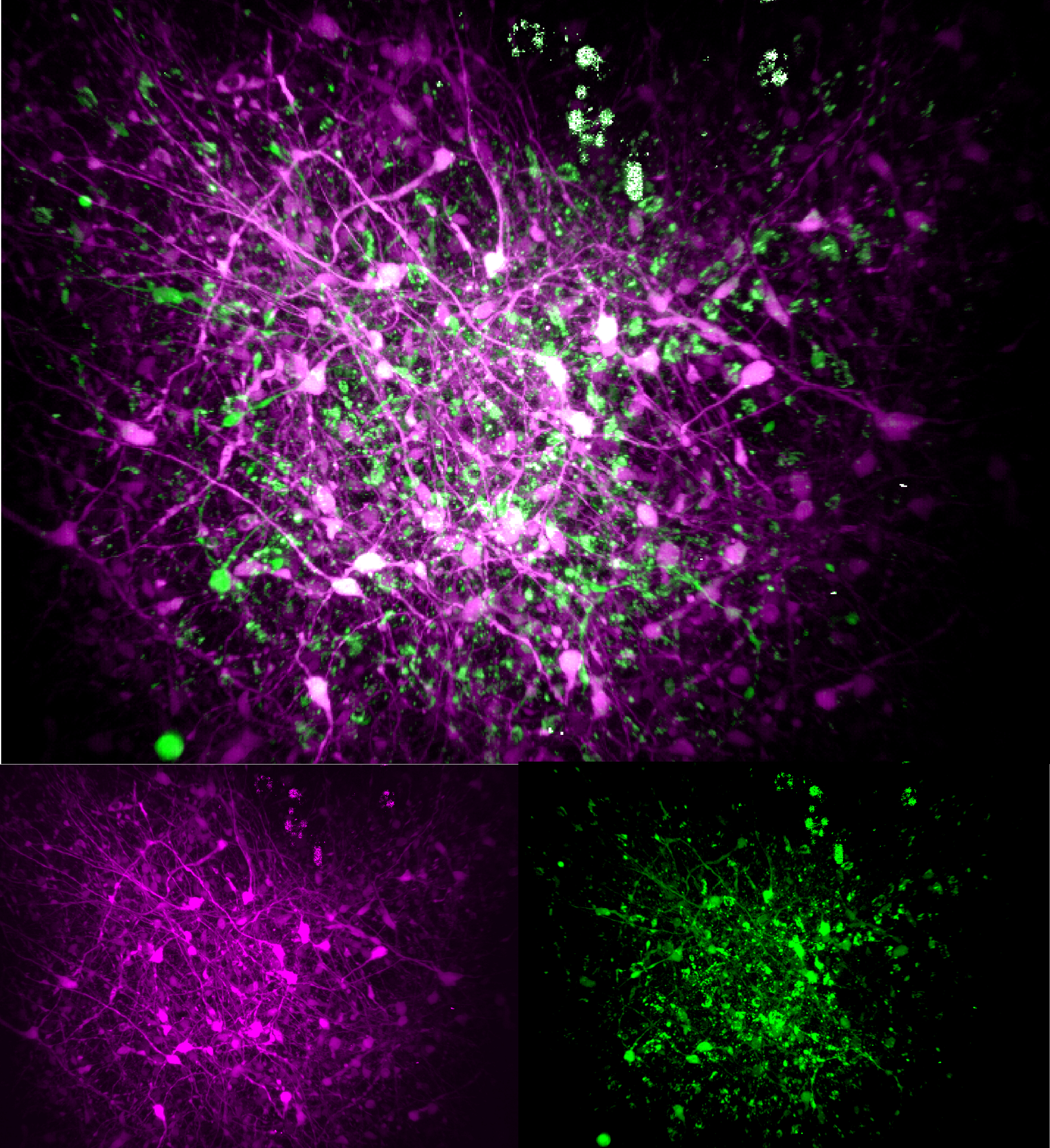
For instance, the scientists observed that the V247X organoids exhibited several structural differences from their controls—they were larger and had different thicknesses of various layers—but the R306C ones were much more like their controls. Organoids harboring either mutation exhibited less developed axon projections from their neurons compared to their control comparators.
Looking at properties of neural activity and connectivity in the organoids, the scientists found some similar deficits across both mutations. Both showed reduced spiking activity and synchronicity between neurons compared to in their controls.
But when the scientists looked at other properties, the organoids started to diverge from each other. In particular, an indication of the efficiency of their network structure called “small-world propensity” (SWP) was decreased in R306C organoids, and increased in V247X ones, compared to controls. This means that both mutations altered the development of typical network structures for information processing, but in different directions.
To ensure that their results were meaningful for Rett syndrome patients, the team collaborated with Charles Nelson at Boston Children’s Hospital, whose team measured EEG in several children with different Rett mutations. Though the sample was small, the researchers measured indications that the SWP property in the EEG readings was altered in the volunteers, much like in the organoids.
Finally, by labeling excitatory neurons to flash in one color and inhibitory neurons to flash in a different color, the scientists were able to see that connectivity between the different neural types differed significantly from controls in the V247X organoids.